整個報告的目錄,合格會有個綠色的對勾,警告WARN是個" ! ",不合格FAIL是個紅色的叉叉。

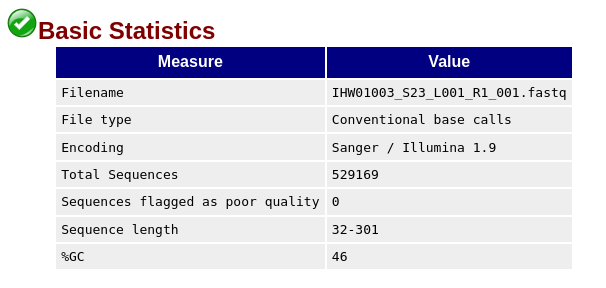
Encoding指測序平台的版本和相應的編碼版本號,這個在計算Phred反推error P的時候有用(Illumina1.9 就是 Phred+33,很重要,1.8以上即為Phred33編碼)
Total sequences: reads數量
Sequence length: 測序長度
%GC(GC含量): 重點關注,可以幫助區別物種,人類42%左右
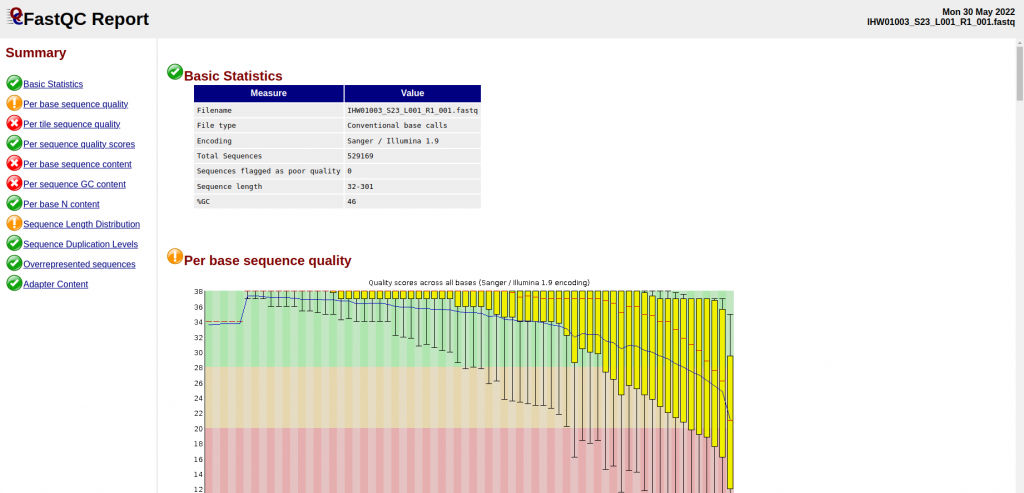
Per base sequence quality
質量高:鹼基質量值都基本都在大於30(質量值的分佈都在綠色背景),而且波動很小,說明質量很穩定
縱軸是質量得分,Q = -10*log10(error P)即20表示1%的錯誤率,30表示0.1%
一般要求此圖中,所有位置的10%分位數大於20,也就是我們常說的Q20過濾
圖中藍色的細線是各個位置的平均值的連線
做read質量值分析的時候,FastQC並不單獨查看具體某一條read中鹼基的質量值,而是將Fastq文件中所有的read數據都綜合起來一起分析。
下圖是read各位置鹼基質量分佈圖
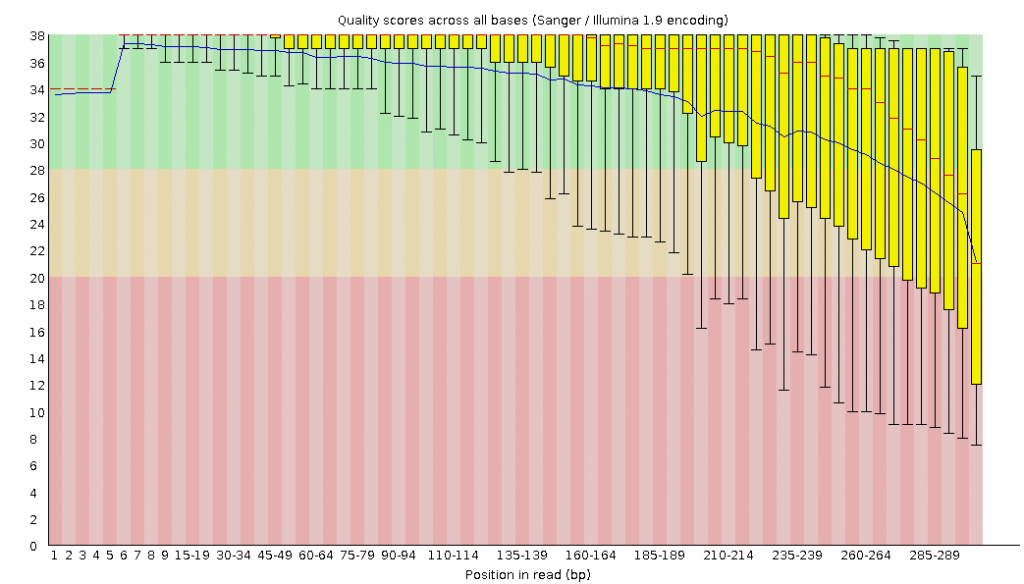
這個圖的橫軸是read上鹼基的位置,縱軸是鹼基質量值。在這個例子中,read的長度是32-301 bp,這應該算是比較長的二代測序序列了。
我們可以看到read上的每一個位置都有一個黃色的箱型圖表示在該位置上所有鹼基的質量分佈情況,在這個圖中我們可以明顯看到,read各個位置上的鹼基質量分佈波動都比較大,特別從第210左右個鹼基往後全部出現了大幅度的波動,而且一些read的鹼基質量值(看黃色箱子)都掉到非常低(紅色)的區域中了,說明這個數據的測序結果中等,有著一些不及格的read。
當然想要最好質量的情況是重新測序,但如果使用這個數據,就要把這些低質量的數據全都去除掉才行,同時還需留意是否還存在其他的問題,但不管如何都一定會丟掉一部分的數據。
鹼基總體質量值分佈,只要大部分都高於20,那麼就比較正常。
關於Q20和Q30的比例是我們衡量測序質量的一個重要指標。這其實也是從這裡來進行體現,一般來說,對於二代測序,最好是達到Q20的鹼基要在95%以上(最差不低於90%),Q30要求大於85%(最差也不要低於80%)
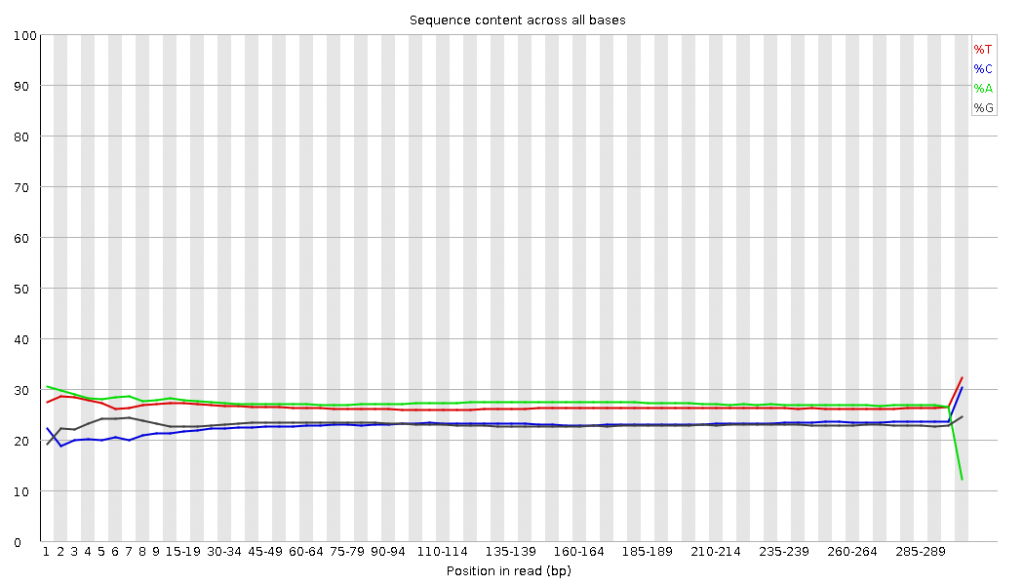
read各個位置上鹼基比例分佈,為了分析鹼基的分離程度。
我們知道AT配對,CG配對,假如測序過程是比較隨機的話(隨機意味著好),那麼在每個位置上A和T比例應該差不多,C和G的比例也應該差不多,如圖所示,兩者之間即使有偏差也不應該太大,最好平均在1%以內,如果過高,除非有合理的原因,比如某些特定的捕獲測序所致,否則都需要注意是不是測序過程有什麼偏差。
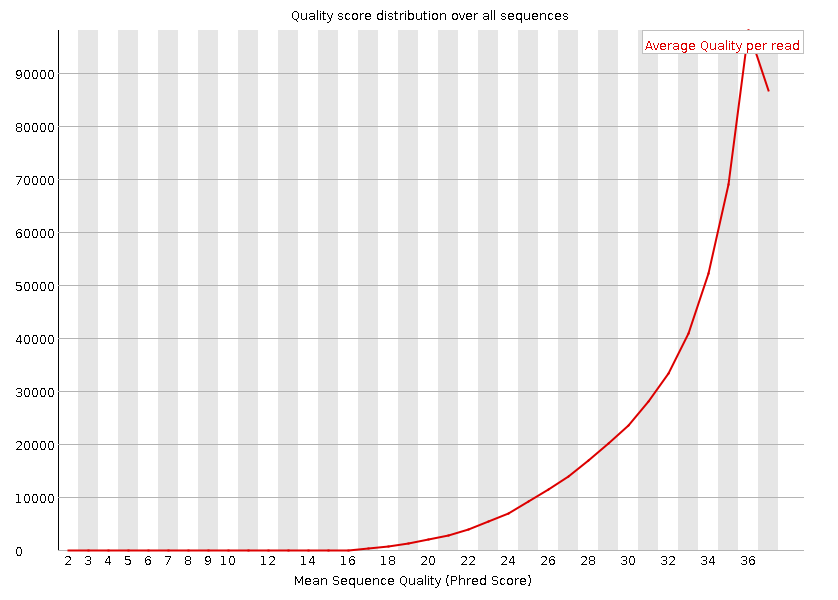
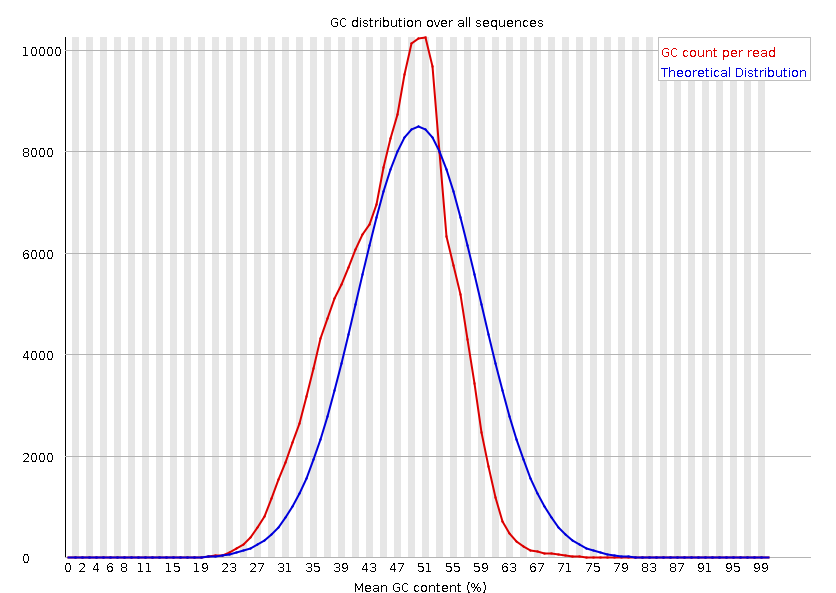
二代定序平台或多或少都存在一定的測序偏向性,我們可以通過查看這個值來協助判斷測序過程是否足夠隨機。對於人類來說,基因組的GC含量一般在40%左右。因此,如果發現GC含量的圖譜明顯偏離這個值那麼說明測序過程存在較高的序列偏向性,結果就是基因組中某些特定區域被反複測序的機率高於平均水平,除了覆蓋度會有偏離之後,將會影響下游的變異檢測和CNV分析。
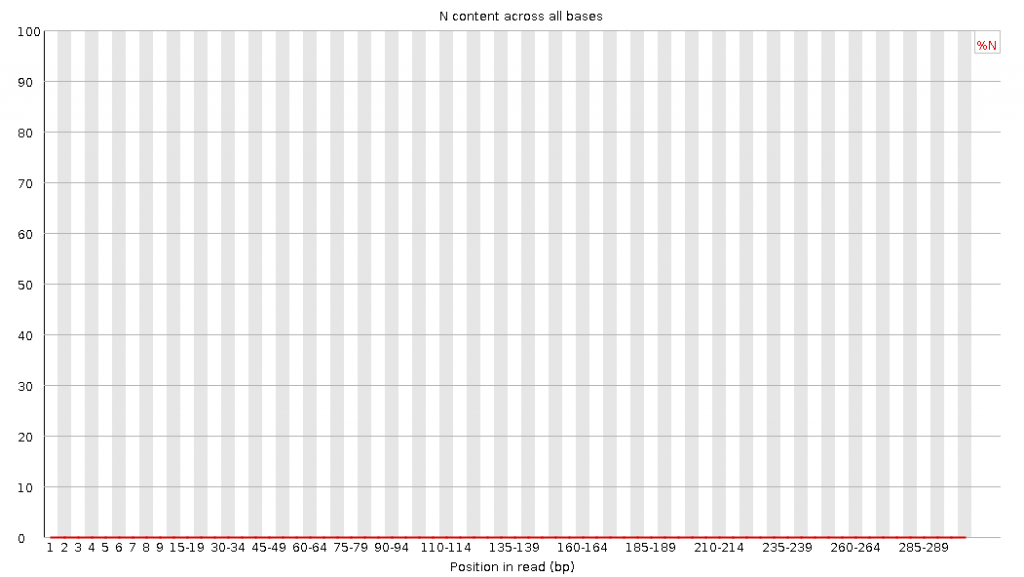
N在測序數據中一般是不應該出現的,如果出現則意味著,測序的光學信號無法被清晰分辨,如果這種情況多的話,往往意味著測序系統或者測序試劑的錯誤。
在sequencing之前需要構建sequencing library,adapter就是在這個時候加上的,其目的一方面是為了能夠結合到flowcell上,另一方面是當有多個樣本同時測序的時候能夠利用adapter進行區分。
當測序read的長度大於被測序的DNA片段時,就會在read的末尾測到這些adapter。
一般的WGS是不會測到這些adapter的,因為構建WGS測序的library序列(插入片段)都比較長,約幾百bp,而read的測序長度都在100bp-150bp這個範圍。
不過在進行一些RNA測序的時候,由於它們的序列本來就比較短,很多只有幾十bp長(特別是miRNA),那麼就很容易會出現read測通的現象,這個時候就會在read的末尾測到這些adapter序列。
通常一般在read前後兩端定序品質都不太穩定,框起來是想表示這個sample中並沒有發現這個現象。
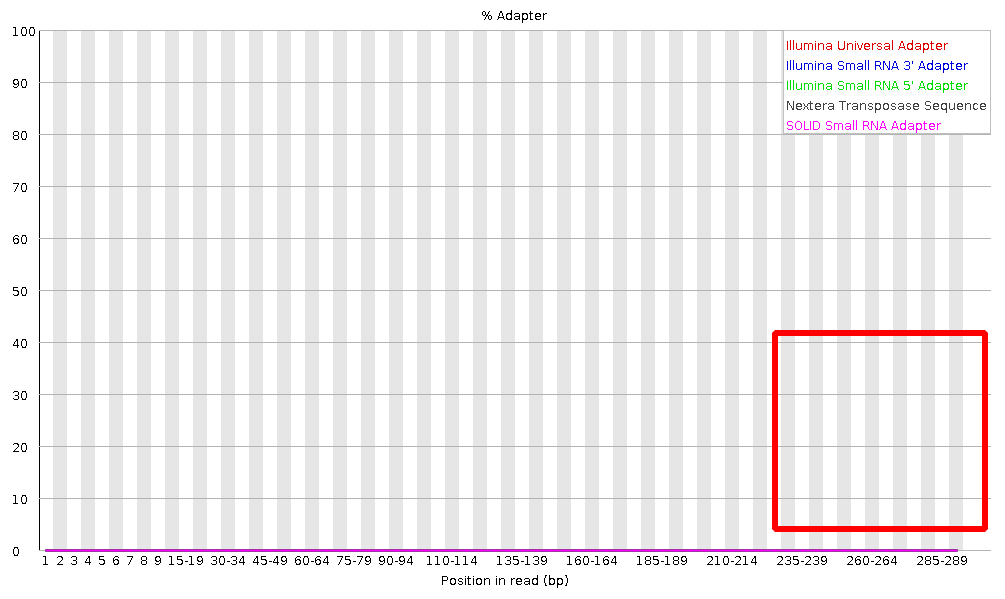
PS. 同學問我平均adapter長度是多少bp呀
我:平均adapter大概20多吧,只是會加上barcode等等標誌,所以長度不等,這個現在可以用一些trim 的軟體切就知道被切掉多長了
EX:fastp

